

Hot topics

Evidence 2013;5(4): e1000039 doi: 10.4470/E1000039
Pubblicato: 29 aprile 2013
Copyright: © 2013 Cartabellotta. Questo è un articolo open-access, distribuito con licenza Creative Commons Attribution, che ne consente l’utilizzo, la distribuzione e la riproduzione su qualsiasi supporto esclusivamente per fini non commerciali, a condizione di riportare sempre autore e citazione originale.

Tutti i professionisti sanitari, medici in particolare, necessitano dei risultati delle sperimentazioni cliniche per scegliere le migliori opzioni terapeutiche: infatti, i trial controllati e randomizzati costituiscono il gold-standard della ricerca per valutare l’efficacia degli interventi sanitari. Attualmente, il “legittimo proprietario” dei dati (i ricercatori o l’industria farmaceutica) può decidere a propria discrezione di non pubblicare i risultati dei trial (bias di pubblicazione) o di farlo in maniera parziale (report selettivo degli outcome). Impedire alla comunità scientifica e ai pazienti di accedere ai risultati integrali di tutti i trial condotti, oltre a distorcere il profilo di efficacia-sicurezza dei trattamenti, aumenta i rischi per i pazienti e consuma preziose risorse. Ad esempio, i medici preferiscono un farmaco nuovo, costoso e dal profilo di sicurezza poco noto, mentre la corrispondente molecola “tradizionale” è più efficace, più sicura e più economica.
Il problema della mancata pubblicazione dei trial è noto da oltre 20 anni, è ampiamente documentato in letteratura, ma non è mai stato risolto in maniera definitiva. Infatti, l’industria farmaceutica tenta di negarne l’esistenza e nessuna delle istituzioni e organizzazioni coinvolte nella ricerca (università , enti di ricerca, agenzie regolatorie, ordini professionali, società scientifiche, comitati etici) si propone per risolverlo definitivamente.
Per tale ragione alcune organizzazioni (Bad Science, Sense About Science, BMJ, James Lind Alliance, Centre for Evidence-based Medicine) hanno promosso la petizione AllTrials per favorire la registrazione di tutti i trial e pubblicarne tutti i risultati. In Italia, AllTrials è stata immediatamente sostenuta dalla Fondazione GIMBE con numerose attività (1), tra cui la traduzione ufficiale dell’editoriale del BMJ che ha lanciato l’iniziativa (2).
1. La dimensione del fenomeno
Una revisione sistematica (3), condotta nel 2010 dal NHS National Institute for Health Research (NIHR), ha documentato due fenomeni inquietanti:
- circa il 50% dei trial condotti sui farmaci, anche se completati, non vengono mai pubblicati per esteso dalle riviste biomediche;
- i trial con risultati positivi hanno il doppio delle probabilitĂ di essere pubblicati rispetto a quelli con risultati negativi.
Questi fenomeni riguardano tutti i trial, indipendentemente dal numero di pazienti arruolati, dal paese in cui viene condotta la sperimentazione clinica, dalla fase di sviluppo di un farmaco o dalla fonte di finanziamento istituzionale o commerciale (4) (figura). PiĂą recentemente, tre studi hanno analizzato il problema della mancata pubblicazione dei trial (box).
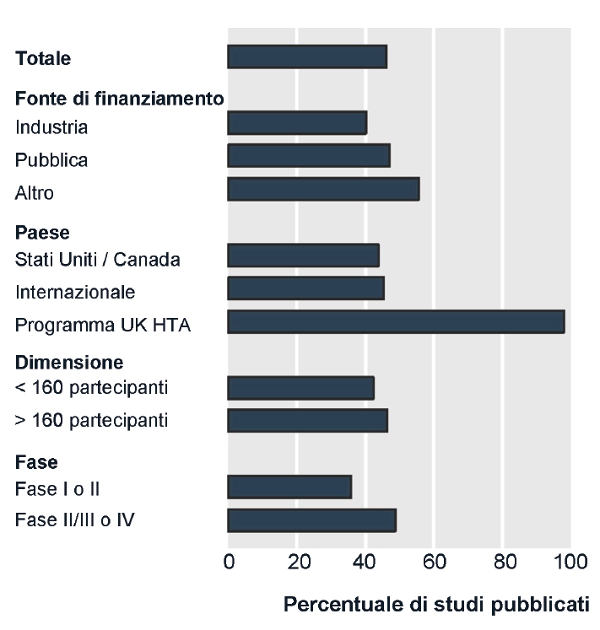
|
Box. Evidenze sulla mancata pubblicazione dei trial Ross JS et coll. hanno dimostrato che di 635 trial clinici conclusi entro il 31 dicembre 2008, solo 294 (46%) sono stati pubblicati da una rivista biomedica indicizzata da Medline entro 30 mesi dalla loro conclusione. Pertanto, nonostante la tempestività delle pubblicazioni sia recentemente migliorata, meno del 50% dei trial finanziati dai National Istitutes of Health (NIH) vengono pubblicati su riviste peer review entro 30 mesi dalla conclusione. Inoltre, dopo una mediana di 51 mesi dalla conclusione, 203 trial (32%) rimangono non pubblicati (5). Bourgeois FT et coll, riportando un tasso di pubblicazione del 66,3%, hanno esplorato il fenomeno del reporting selettivo degli outcome (outcome reporting bias). I trial sponsorizzati dall’industria riportano outcome positivi nell’85,4% delle pubblicazioni, rispetto al 50% dei trial con finanziamenti governativi e al 71,9% di quelli finanziati da organizzazioni no-profit o non federali (p < 0.001). La percentuale di trial pubblicati entro 24 mesi dalla conclusione varia dal 32,4% per quelli finanziati dall’industria al 56,2% per quelli finanziati da organizzazioni no-profit o non federali senza contributi dall’industria (p = 0.005) (6). Zarin DA et coll., hanno analizzato i record pubblicamente disponibili sul registro ClinicalTrials.gov nel periodo settembre 2009 - settembre 2010. Su un campione di 150 trial, 78 (52%) erano stati pubblicati entro due anni dalla loro registrazione. Gli autori dimostrano che ClinicalTrials.gov permette di accedere ai risultati di studi altrimenti non disponibili al pubblico e di esaminare vari aspetti sia dei trial in corso, sia di quelli già completati. Ovviamente, l’utilità di questo registro è strettamente legata all’input di dati accurati e informativi” (7). |
2. Iniziative internazionali
Due rilevanti iniziative vengono continuamente citate a riprova del fatto che il problema è ormai risolto:
- la registrazione dei trial quale requisito per la pubblicazione;
- la normativa della Food and Drug Administration (FDA) che prevede la pubblicazione dei risultati dei trial sul registro ClinicalTrials.gov entro un anno dalla loro conclusione.
2.1. Registrazione dei trial quale requisito per la pubblicazione
Da oltre 15 anni i ricercatori sono incoraggiati ad inserire i trial in un registro pubblicamente accessibile: in questo modo, anche se il reporting dei risultati non è garantito, è possibile verificare se gli studi completati siano stati, o meno, pubblicati.
Visto che, nonostante varie iniziative di sensibilizzazione, la registrazione dei trial continuava a dipendere dalla volontà dei ricercatori, nel 2005 l’International Committee of Medical Journal Editors (ICMJE) ha pubblicato uno statement dichiarando che sarebbero stati pubblicati solo i trial registrati al momento del loro inizio. Nonostante numerose riviste di tutto il mondo aderenti all’ICMJE sostengano formalmente questa lodevole iniziativa, non è mai stato verificato l’impatto reale dello statement dell’ICMJE. Peraltro nel 2009 Mathieu S et coll. hanno dimostrato che il 50% dei trial pubblicati successivamente allo statement dell’ICMJE non erano stati adeguatamente registrati e il 25% non era mai stato registrato (8).
2.1.1. Il registro dell’European Medicines Agency
Nel 2001 e nel 2004 è stato richiesto all’European Medicines Agency (EMA) di realizzare un registro dei trial, al fine di allineare gli standard europei a quelli USA, che dispongono di ClinicalTrials.gov. Il 10 marzo 2011 l’EMA ha reso disponibile un registro pubblico (www.clinicaltrialsregister.eu), precisando che le informazioni relative al periodo 2004-2011 sono “storiche” e pertanto non affidabili. Pertanto, se è implicito che un registro debba rendere accessibili i dettagli di ciascun trial, in quello dell’EMA di fatto non sono consultabili i dettagli relativi a migliaia di trial archiviati. Inoltre, a differenza di Clinicaltrials.gov e indipendentemente dalla volontà dei ricercatori, le informazioni sui trial di fase 1 vengono mantenute segrete, a dispetto della necessità etica e scientifica di rendere pubblica anche questa fase. Infine, nel caso di trial archiviati in diversi registri, quello dell’EMA spesso contiene errori e incongruenze rispetto ad altri.
Nonostante queste ambiguità il registro dell’EMA è stato inserito nell’elenco dei registri primari della International Clinical Trials Registry Platform (ICTRP) (9), iniziativa dell’Organizzazione Mondiale della Sanità finalizzata alla registrazione di tutti i trial quale “responsabilità scientifica, etica e morale”.
2.2. La normativa della Food and Drugs Administration
L’Amendment Act 2007 della FDA prevede che, per tutti i trial farmacologici condotti negli Stati Uniti (o, nel caso di trial multicentrici, con almeno un centro in USA) i risultati devono essere pubblicati nel registro ClinicalTrials.gov entro un anno dalla conclusione della sperimentazione. Di fatto, non sono mai state attuate verifiche sull’aderenza a questa disposizione, né i dati sono pubblicamente accessibili.
Nonostante uno studio indipendente abbia dimostrato che solo il 20% dei trial soddisfa questo requisito (10), alle aziende farmaceutiche o ai ricercatori inadempienti non è mai stata comminata alcuna sanzione, che peraltro ammonterebbe a “soli” $ 10.000,00/die. Ben poca cosa per aziende che incassano decine di miliardi di dollari l’anno e per le quali il fatturato relativo a un farmaco di largo consumo può superare i 5 miliardi di dollari.
In ogni caso, anche se attuata, la normativa della FDA non avrebbe risolto il problema: infatti, l’obbligo di pubblicazione riguarda i trial avviati dopo il 2009, mentre i medici si basano generalmente su evidenze da studi precedenti. Peraltro, la normativa non si estende ai trial condotti al di fuori degli USA, sempre più frequenti da quando le contract research organisations (CRO) hanno intuito che esistono paesi più “tolleranti”, quali Brasile, Russia, India, Cina, etc.
3. Tamiflu e Relenza: il mancato accesso ai report integrali degli studi clinici
Il Tamiflu (oseltamivir) è un farmaco per cui i sistemi sanitari hanno speso milioni di euro/dollari/sterline in scorte di magazzino, in particolare a seguito della pandemia H1N1, frettolosamente paventata dall’OMS. Delle 1.200.000 dosi vendute in Gran Bretagna nella stagione influenzale 2009-2010, almeno la metà è rimasta inutilizzata, con uno spreco di 7,8 milioni di sterline (11).
Per la rilevanza della ricerca condotta dal Cochrane Acute Respiratory Infections Group, quello del Tamiflu è probabilmente il caso meglio documentato sull’occultamento dei dati dei trial e sulla mancata trasparenza da parte dell’industria, ma indubbiamente non si tratta di un caso isolato. Considerato che per diversi studi sul Tamiflu sono disponibili solo brevi sintesi per il pubblico, che non contengono dettagli rilevanti per valutarne rigore metodologico e affidabilità , i ricercatori della Cochrane Collaboration hanno richiesto alla Roche, azienda produttrice del farmaco, i report completi degli studi clinici. Nel dicembre 2009 la Roche si è impegnata per iscritto a condividere questi documenti: in realtà , oltre a non avere onorato l’impegno, ha proposto di convocare un comitato per esaminare la questione, un atteggiamento che non va certo nella direzione della trasparenza (12,13).
A differenza della Roche, la GlaxoSmithKline (GSK) ha dichiarato che condividerà con il Cochrane Acute Respiratory Infections Group i dati sul Relenza (zanamivir), il suo farmaco antinfluenzale. Dopo una lunga negoziazione, con spreco di giornate lavorative e impiego di ricercatori senior e legali all’Università di Oxford, la Cochrane Collaboration ha ricevuto un DVD contenente una versione riveduta e corretta del report del trial. Le “censure” non erano limitate ai dati identificativi dei pazienti, ma si estendevano ai loro numeri di identificazione (assegnati da GSK per aggregare gli outcome), rendendo impossibile un’analisi adeguata. Pertanto, si continua a discutere per individuare esattamente cosa serve alla Cochrane Collaboration e che cosa GSK è disposta a fornire.
4. I responsabili dell’occultamento dei trial: non solo industria e ricercatori
Gran parte delle critiche si sono legittimamente concentrate sulle sperimentazioni finanziate dall’industria, sia perché questa sponsorizza la maggior parte dei trial, sia perché esistono consistenti e inequivocabili evidenze di under-reporting da parte delle compagnie farmaceutiche e biomedicali. Tuttavia il fenomeno non è confinato ai trial finanziati da sponsor commerciali, tanto che i primi esempi di mancata pubblicazione di risultati negativi vengono proprio dal mondo accademico. Inoltre:
- le UniversitĂ e i centri di ricerca, nei contratti stipulati con le aziende che finanziano i trial, non impongono la pubblicazione dei risultati, indipendentemente dal fatto che soddisfino le esigenze dello sponsor;
- i comitati etici che approvano i progetti di ricerca non sono in grado di proteggere adeguatamente i pazienti, poiché non insistono sulla necessità della pubblicazione dei risultati, né effettuano verifiche sulla pubblicazione dei trial già approvati;
- gli ordini professionali e le società scientifiche non hanno mai affrontato il problema, né hanno mai dichiarato apertamente che non pubblicare, e quindi occultare, i risultati dei trial clinici costituisce una frode scientifica.
È fondamentale sottolineare che la mancata pubblicazione dei risultati di un trial costituisce una gravissima violazione etica. Infatti, i partecipanti arruolati in un trial forniscono il proprio consenso informato, sottoponendosi a disagi, inconvenienti e rischi, nella certezza che la loro partecipazione contribuirĂ a migliorare le conoscenze scientifiche. Occultare i risultati di un trial equivale ad ingannare i partecipanti, violando il patto siglato con il consenso informato.
5. Le riviste scientifiche: un falso ostacolo
Ricercatori e industria puntano il dito contro le riviste scientifiche per la scarsa disponibilità a pubblicare i trial con risultati negativi. In realtà questo problema, attualmente di modesta entità , è risolto da oltre dieci anni. Infatti, la revisione sistematica NHS NIHR (3) ha dimostrato che le riviste non costituiscono un ostacolo alla pubblicazione. Peraltro, con la crescita esponenziale delle riviste open access, dove il business non è più legato alla vendita di abbonamenti, pubblicità e ristampe, numerose riviste pubblicano i trial indipendentemente dal fatto che i loro risultati siano positivi e permettono a chiunque di leggere e valutare gli articoli in maniera gratuita.
6. La soluzione? Coinvolgere tutti gli stakeholders
Anche se può sembrare paradossale, oggi non è obbligatorio per legge rendere disponibili i risultati dei trial condotti sui farmaci di uso corrente. Infatti, la normativa impone di riportare eventuali eventi avversi, di monitorare le sperimentazioni, di garantire la “buona pratica clinica” nella conduzione dei trial, ma non affronta le distorsioni conseguenti sia al reporting incompleto, sia alla mancata pubblicazione dei risultati.
La petizione AllTrials chiede che “tutti i risultati di tutti i trial pregressi e futuri condotti su tutti gli interventi sanitari siano resi disponibili” per garantire decisioni informate sugli interventi terapeutici. E’ indubbio che tutti gli stakeholders, mettendo da parte i propri interessi di categoria, dovrebbero essere coinvolti per risolvere definitivamente il problema.
- Le Istituzioni potrebbero applicare un approccio piĂą rigoroso. Ad esempio il SSN dovrebbe rimborsare un trattamento solo se tutti i dati di tutti i trial che lo riguardano sono pubblicamente accessibili; oppure inserire in prontuario solo i trattamenti delle aziende farmaceutiche che rendono pubblicamente disponibili i risultati di tutti i trial condotti su tutti i farmaci da loro prodotti.
- I comitati etici dovrebbero giocare un ruolo attivo nel prevenire il bias di pubblicazione, effettuando un monitoraggio pubblico dei protocolli approvati. Inoltre, potrebbero verificare l’esistenza di pubblicazioni dei trial precedenti dei ricercatori prima di autorizzare ulteriori progetti di ricerca.
- Le riviste potrebbero attuare una politica simile a quella del BMJ che prende in considerazione per la pubblicazione i trial su farmaci e dispositivi medici solo se gli autori si impegnano a rendere disponibili in forma anonima i dati sui pazienti, a seguito di richiesta motivata.
- Le università e i centri di ricerca non dovrebbero sottoscrivere contratti di sponsorizzazione dove l’industria detiene la proprietà dei dati e, di conseguenza, decide se pubblicare o meno i trial.
- I pazienti dovrebbero boicottare i trial che non garantiscono la pubblicazione (14) e partecipare solo a quelli che, oltre ad avere un protocollo registrato, pubblicamente accessibile e che fa riferimento a revisioni sistematiche delle evidenze disponibili che ne giustificano la necessitĂ , rilasciano una garanzia scritta che i risultati completi dello studio saranno pubblicati e inviati a tutti i partecipanti che lo desiderano (2).
Last but not least, la Commissione Europea ha recentemente elaborato la “Proposta di Regolamento del Parlamento Europeo e del Consiglio concernente la sperimentazione clinica di medicinali per uso umano, e che abroga la direttiva 2001/20/CE (15)”. Tale proposta, estremamente carente su vari aspetti metodologici (16), non solo ignora il problema della mancata pubblicazione dei trial e la necessità di registrarli, ma addirittura prevede che il protocollo di una sperimentazione clinica descriva “la politica di pubblicazione”, lasciando intendere che registrazione e pubblicazione di un trial non rappresentano un obbligo scientifico, etico e morale, ma solo “optional opportunistici”.
Il testo è in fase di revisione da parte delle Autorità Regolatorie degli Stati Membri: nel nostro Paese, l’Agenzia Italiana per il Farmaco (AIFA), al fine di raccogliere i commenti e le proposte dei diversi stakeholders nazionali, funge da “collettore nazionale” per la consultazione pubblica che si concluderà il 3 giugno 2013.
Contributo degli Autori
-Disclosure dei conflitti di interesse
AC è il Presidente del GIMBE, organizzazione no-profit che svolge attività di formazione e ricerca sugli argomenti trattati nell’articoloIndirizzo per la corrispondenza
nino.cartabellotta@gimbe.orgProvenienza
Non commissionato, non sottoposto a peer-reviewFonti di finanziamento
NessunaApprovazione comitato etico
Non richiestaPagina aggiornata il 29/aprile/2013